publications
publications in reversed chronological order. First authorships marked with asterisk (*)
2026
-
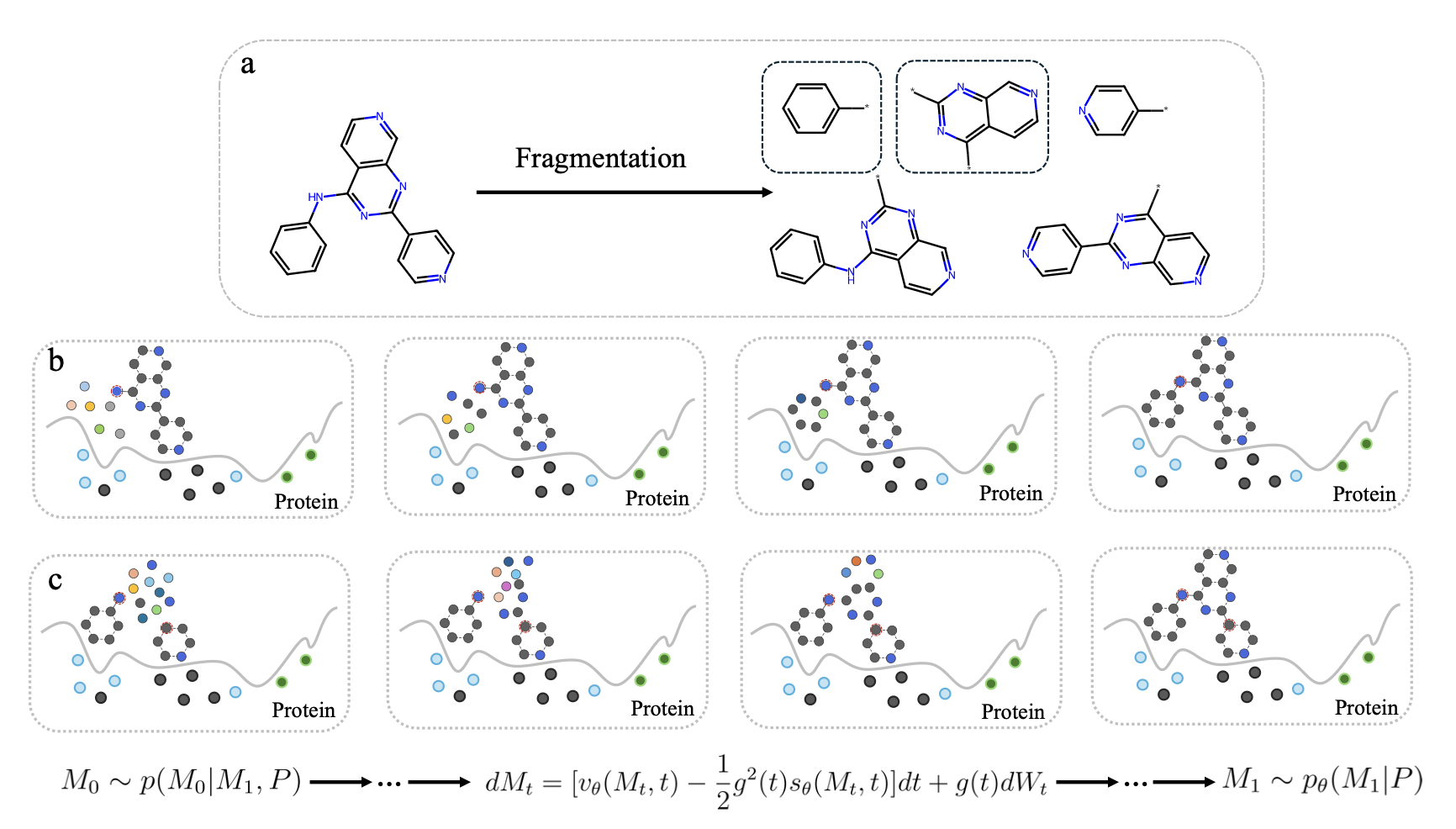 Coupled fragment-based generative modeling with stochastic interpolantsDigital Discovery Mar 2026
Coupled fragment-based generative modeling with stochastic interpolantsDigital Discovery Mar 2026Fragment-based drug design (FBDD) has become a key approach in structure-based drug discovery, allowing researchers to systematically develop molecular fragments into potent ligands. Although recent generative AI models, such as diffusion-based approaches, show great potential for designing new molecules, applying them to fragment-based methods faces challenges due to mismatches between training and inference procedures, as well as computational limitations. In this work, we develop a generative model based on stochastic interpolants that unify diffusion and flow matching paradigms, learning to create fragments through conditional training on molecular substructures. Our experiments show that models trained with explicit fragment-based conditioning perform much better than unconditional models that are adapted for fragment completion tasks. We compare diffusion models with flow matching models using identical backbone architectures and find that flow matching delivers better convergence and produces higher-quality 3D molecular poses with reduced strain energies, all while needing fewer computational steps. We test our method on standard benchmark datasets and examine different fragmentation strategies, finding that the choice of fragmentation algorithm plays an important role in model performance. Through a detailed case study on an internal PLK3 inhibitor structure, we demonstrate that our approach can generate new fragments that show computationally favorable docking scores and binding energy estimates competitive with tested internal Pfizer compounds, while also exploring regions of chemical space that go beyond existing fragment libraries. These findings establish flow matching within the stochastic interpolants framework as a promising approach for fragment-based drug design, providing both improved computational efficiency and better molecular quality for structure-based optimization.
2025
-
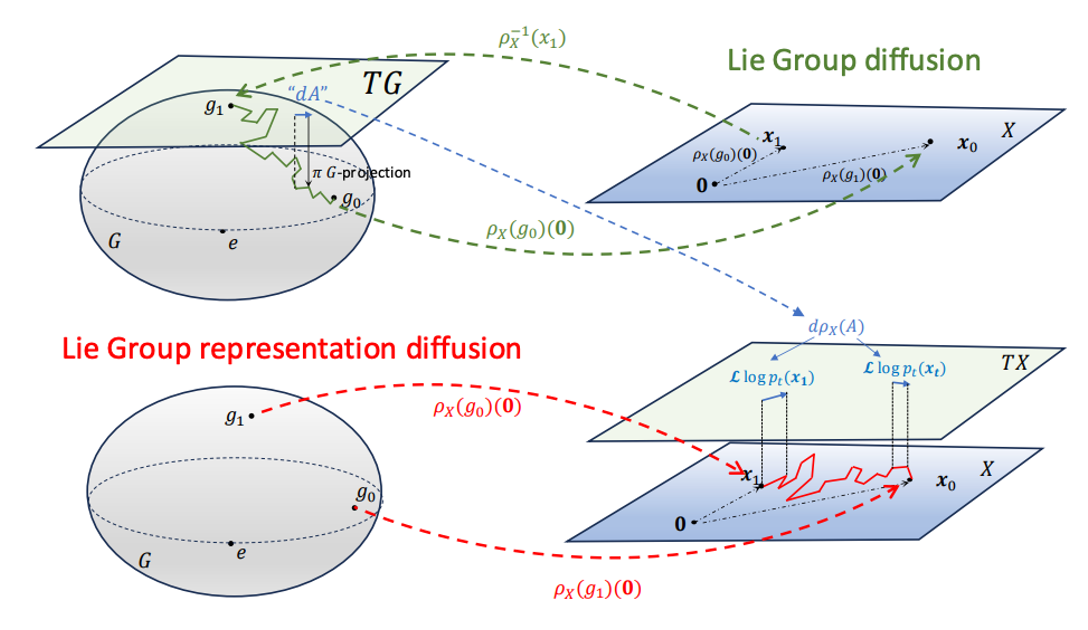 Diffusion Generative Modeling on Lie Group RepresentationsMarco Bertolini*, Tuan Le*, and Djork-Arné ClevertIn The Thirty-ninth Annual Conference on Neural Information Processing Systems Dec 2025
Diffusion Generative Modeling on Lie Group RepresentationsMarco Bertolini*, Tuan Le*, and Djork-Arné ClevertIn The Thirty-ninth Annual Conference on Neural Information Processing Systems Dec 2025We introduce a novel class of score-based diffusion processes that operate directly in the representation space of Lie groups. Leveraging the framework of Generalized Score Matching, we derive a class of Langevin dynamics that decomposes as a direct sum of Lie algebra representations, enabling the modeling of any target distribution on any (non-Abelian) Lie group. Standard score-matching emerges as a special case of our framework when the Lie group is the translation group T(N). We prove that our generalized generative processes arise as solutions to a new class of paired stochastic differential equations (SDEs), introduced here for the first time. We validate our approach through experiments on diverse data types, demonstrating its effectiveness in real-world applications such as SO(3)-guided molecular conformer generation and modeling ligand-specific global SE(3) transformations for molecular docking, showing improvement in comparison to Riemannian diffusion on the group itself. We show that an appropriate choice of Lie group enhances learning efficiency by reducing the effective dimensionality of the trajectory space and enables the modeling of transitions between complex data distributions.
-
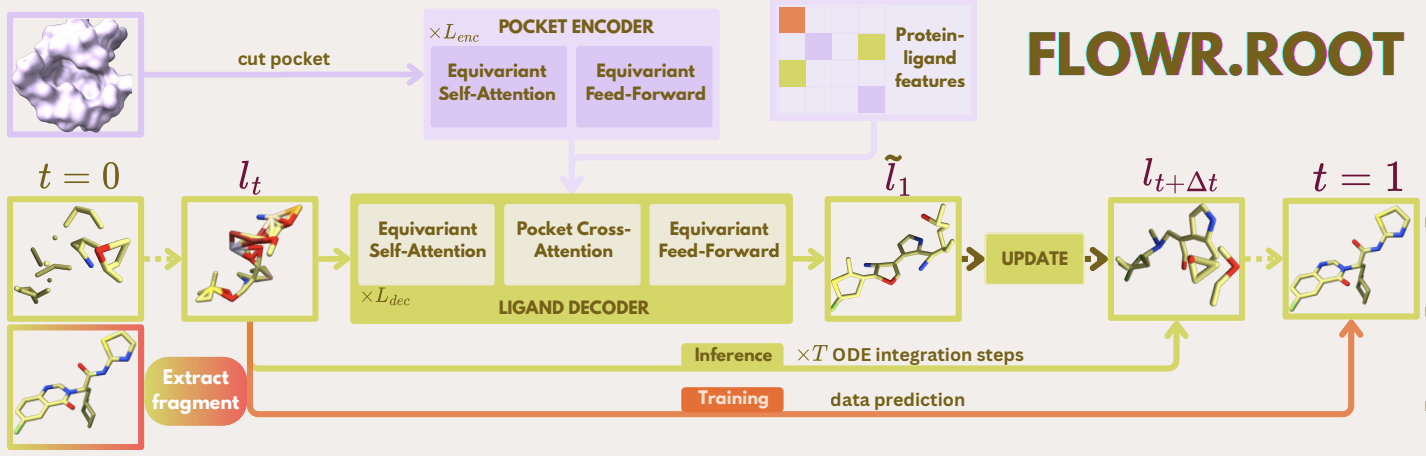 FLOWR.root: A flow matching based foundation model for joint
FLOWR.root: A flow matching based foundation model for joint
multi-purpose structure-aware 3D ligand generation and affinity predictionJulian Cremer*, Tuan Le, Mohammad M. Ghahremanpour, Emilia Sługocka, and 2 more authorsDec 2025We present FLOWR:root, an equivariant flow-matching model for pocket-aware 3D ligand generation with joint binding affinity prediction and confidence estimation. The model supports de novo generation, pharmacophore-conditional sampling, fragment elaboration, and multi-endpoint affinity prediction (pIC50, pKi, pKd, pEC50). Training combines large-scale ligand libraries with mixed-fidelity protein-ligand complexes, followed by refinement on curated co-crystal datasets and parameter-efficient finetuning for project-specific adaptation. FLOWR:root achieves state-of-the-art performance in unconditional 3D molecule generation and pocket-conditional ligand design, producing geometrically realistic, low-strain structures. The integrated affinity prediction module demonstrates superior accuracy on the SPINDR test set and outperforms recent models on the Schrodinger FEP+/OpenFE benchmark with substantial speed advantages. As a foundation model, FLOWR:root requires finetuning on project-specific datasets to account for unseen structure-activity landscapes, yielding strong correlation with experimental data. Joint generation and affinity prediction enable inference-time scaling through importance sampling, steering molecular design toward higher-affinity compounds. Case studies validate this: selective CK2α ligand generation against CLK3 shows significant correlation between predicted and quantum-mechanical binding energies, while ERα and TYK2 scaffold elaboration demonstrates strong agreement with QM calculations. By integrating structure-aware generation, affinity estimation, and property-guided sampling, FLOWR:root provides a comprehensive foundation for structure-based drug design spanning hit identification through lead optimization.
-
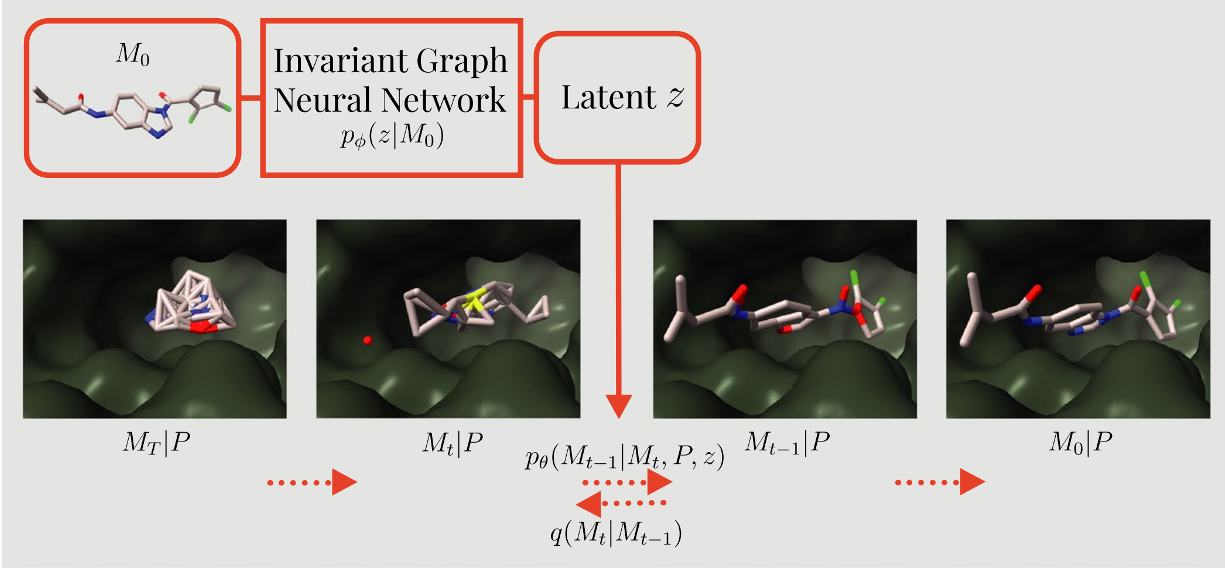 Equivariant diffusion for structure-based de novo ligand generation with latent-conditioningJournal of Cheminformatics May 2025
Equivariant diffusion for structure-based de novo ligand generation with latent-conditioningJournal of Cheminformatics May 2025We introduce PoLiGenX, a novel generative model for de novo ligand design that employs latent-conditioned, target-aware equivariant diffusion. Our approach leverages the conditioning of the ligand generation process on reference molecules located within a specific protein pocket. By doing so, PoLiGenX generates shape-similar ligands that are adapted to the target pocket, enabling effective applications in target-aware hit expansion and hit optimization. Our experimental results underscore the efficacy of PoLiGenX in advancing ligand design. Notably, docking analyses reveal that the ligands generated by PoLiGenX show enhanced binding affinities relative to their reference molecules, all while retaining a similar molecular shape, but also retaining better poses with lower strain energies and less steric clashes. Furthermore, the model promotes substantial chemical diversity, facilitating the exploration of broader and more varied chemical spaces. Importantly, the generated ligands were assessed for drug-likeness using Lipinski’s rule of five, demonstrating superior adherence to drug-likeness criteria compared to the reference dataset. This work represents a step forward in the controlled and precise generation of therapeutically relevant de novo ligands tailored for specific protein targets, contributing to progress in computational drug discovery and ligand design.
2024
-
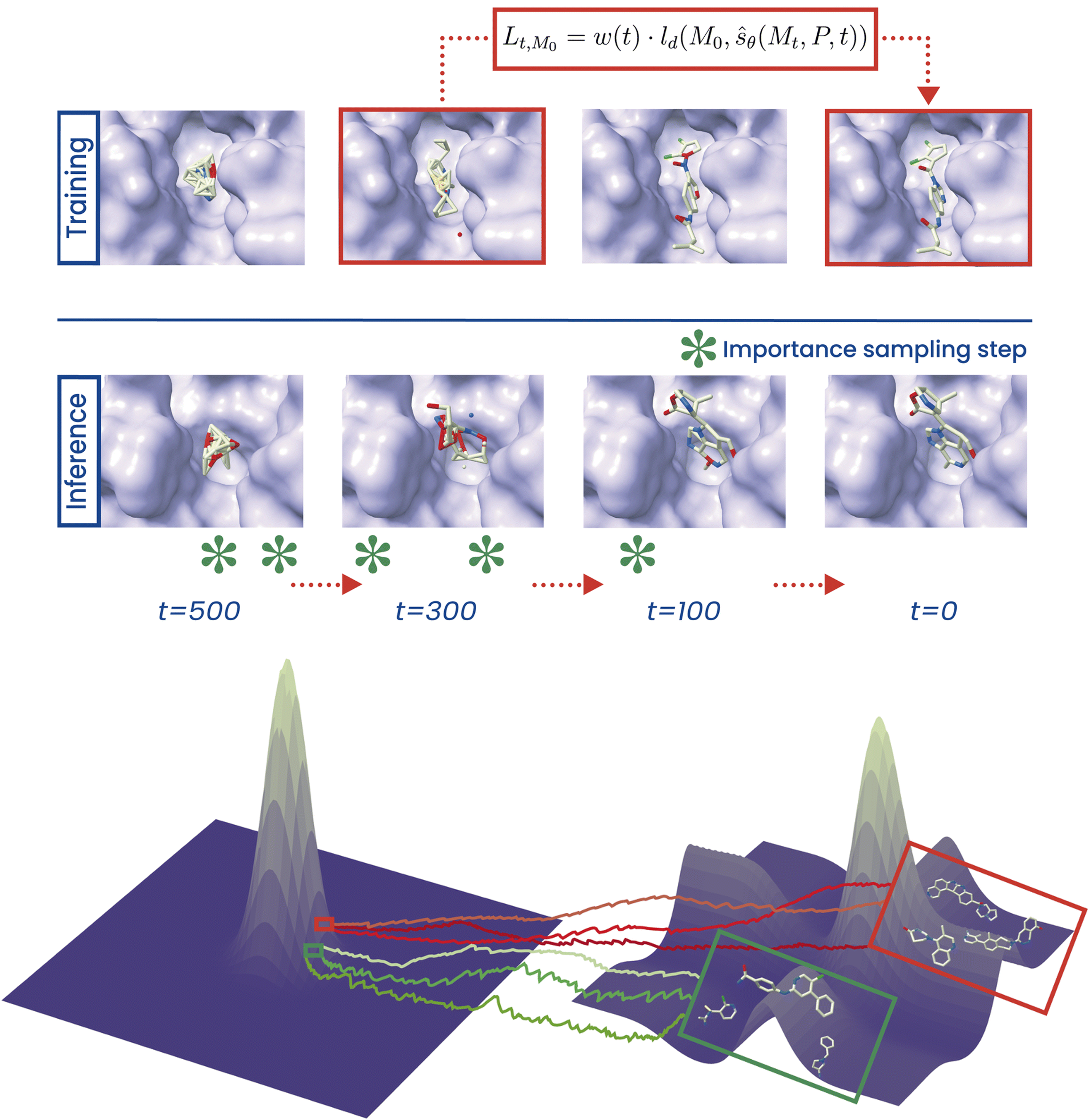 PILOT: equivariant diffusion for pocket-conditioned de novo ligand generation with multi-objective guidance via importance samplingChem. Sci. May 2024
PILOT: equivariant diffusion for pocket-conditioned de novo ligand generation with multi-objective guidance via importance samplingChem. Sci. May 2024The generation of ligands that both are tailored to a given protein pocket and exhibit a range of desired chemical properties is a major challenge in structure-based drug design. Here, we propose an in silico approach for the de novo generation of 3D ligand structures using the equivariant diffusion model PILOT, combining pocket conditioning with a large-scale pre-training and property guidance. Its multi-objective trajectory-based importance sampling strategy is designed to direct the model towards molecules that not only exhibit desired characteristics such as increased binding affinity for a given protein pocket but also maintains high synthetic accessibility. This ensures the practicality of sampled molecules, thus maximizing their potential for the drug discovery pipeline. PILOT significantly outperforms existing methods across various metrics on the common benchmark dataset CrossDocked2020. Moreover, we employ PILOT to generate novel ligands for unseen protein pockets from the Kinodata-3D dataset, which encompasses a substantial portion of the human kinome. The generated structures exhibit predicted IC50 values indicative of potent biological activity, which highlights the potential of PILOT as a powerful tool for structure-based drug design.
-
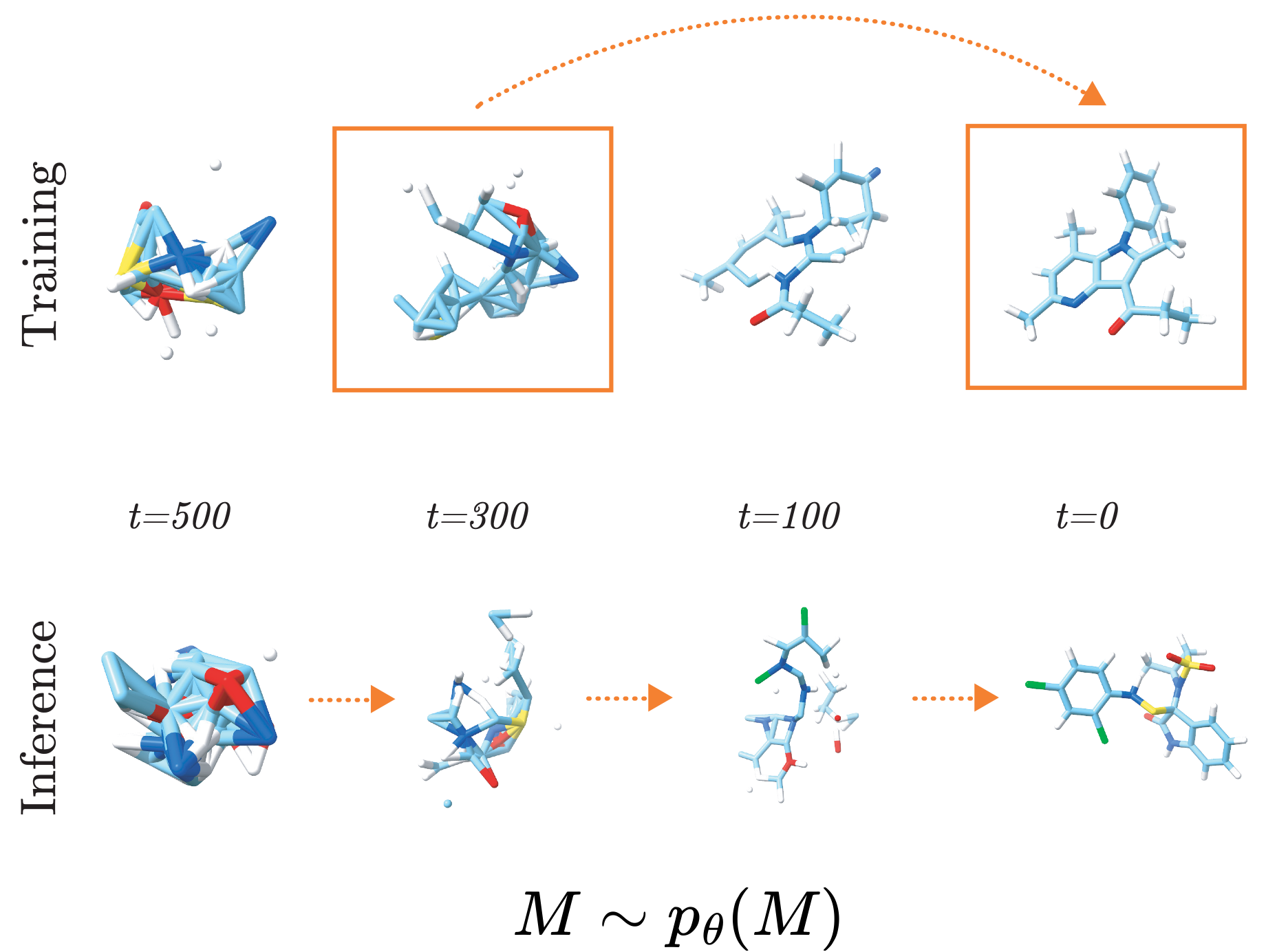 Navigating the Design Space of Equivariant Diffusion-Based Generative Models for De Novo 3D Molecule GenerationIn The Twelfth International Conference on Learning Representations May 2024
Navigating the Design Space of Equivariant Diffusion-Based Generative Models for De Novo 3D Molecule GenerationIn The Twelfth International Conference on Learning Representations May 2024Deep generative diffusion models are a promising avenue for 3D de novo molecular design in materials science and drug discovery. However, their utility is still limited by suboptimal performance on large molecular structures and limited training data. To address this gap, we explore the design space of E(3)-equivariant diffusion models, focusing on previously unexplored areas. Our extensive comparative analysis evaluates the interplay between continuous and discrete state spaces. From this investigation, we present the EQGAT-diff model, which consistently outperforms established models for the QM9 and GEOM-Drugs datasets. Significantly, EQGAT-diff takes continuous atom positions, while chemical elements and bond types are categorical and uses time-dependent loss weighting, substantially increasing training convergence, the quality of generated samples, and inference time. We also showcase that including chemically motivated additional features like hybridization states in the diffusion process enhances the validity of generated molecules. To further strengthen the applicability of diffusion models to limited training data, we investigate the transferability of EQGAT-diff trained on the large PubChem3D dataset with implicit hydrogen atoms to target different data distributions. Fine-tuning EQGAT-diff for just a few iterations shows an efficient distribution shift, further improving performance throughout data sets. Finally, we test our model on the Crossdocked data set for structure-based de novo ligand generation, underlining the importance of our findings showing state-of-the-art performance on Vina docking scores.
2023
-
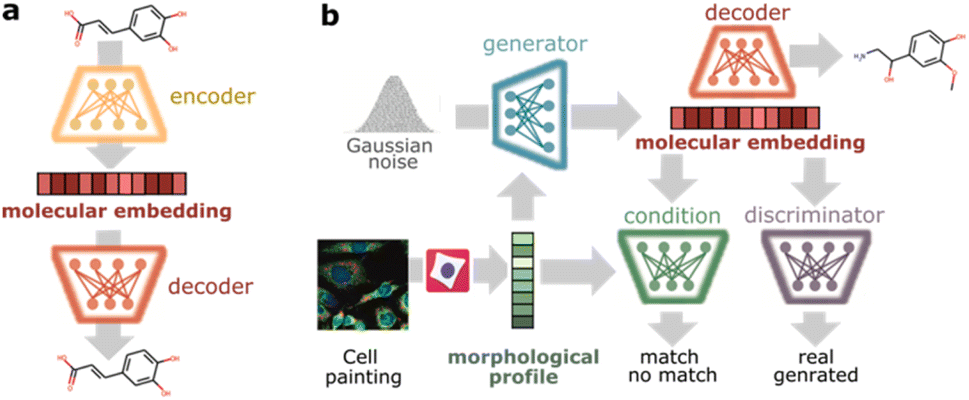 Cell morphology-guided de novo hit design by conditioning GANs
Cell morphology-guided de novo hit design by conditioning GANs
on phenotypic image featuresPaula A. Marin Zapata*, Oscar Méndez-Lucio*, Tuan Le, Carsten Jörn Beese, and 3 more authorsDigital Discovery May 2023Developing novel bioactive molecules is time-consuming, costly and rarely successful. As a mitigation strategy, we utilize, for the first time, cellular morphology to directly guide the de novo design of small molecules. We trained a conditional generative adversarial network on a set of 30 000 compounds using their cell painting morphological profiles as conditioning. Our model was able to learn chemistry-morphology relationships and influence the generated chemical space according to the morphological profile. We provide evidence for the targeted generation of known agonists when conditioning on gene overexpression profiles, even though no information on biological targets was used during training. Based on a target-agnostic readout, our approach facilitates knowledge transfer between biological pathways and can be used to design bioactives for many targets under one unified framework. Prospective application of this proof-of-concept to larger chemical spaces promises great potential for hit generation in drug and phytopharmaceutical discovery and chemical safety.
2022
-
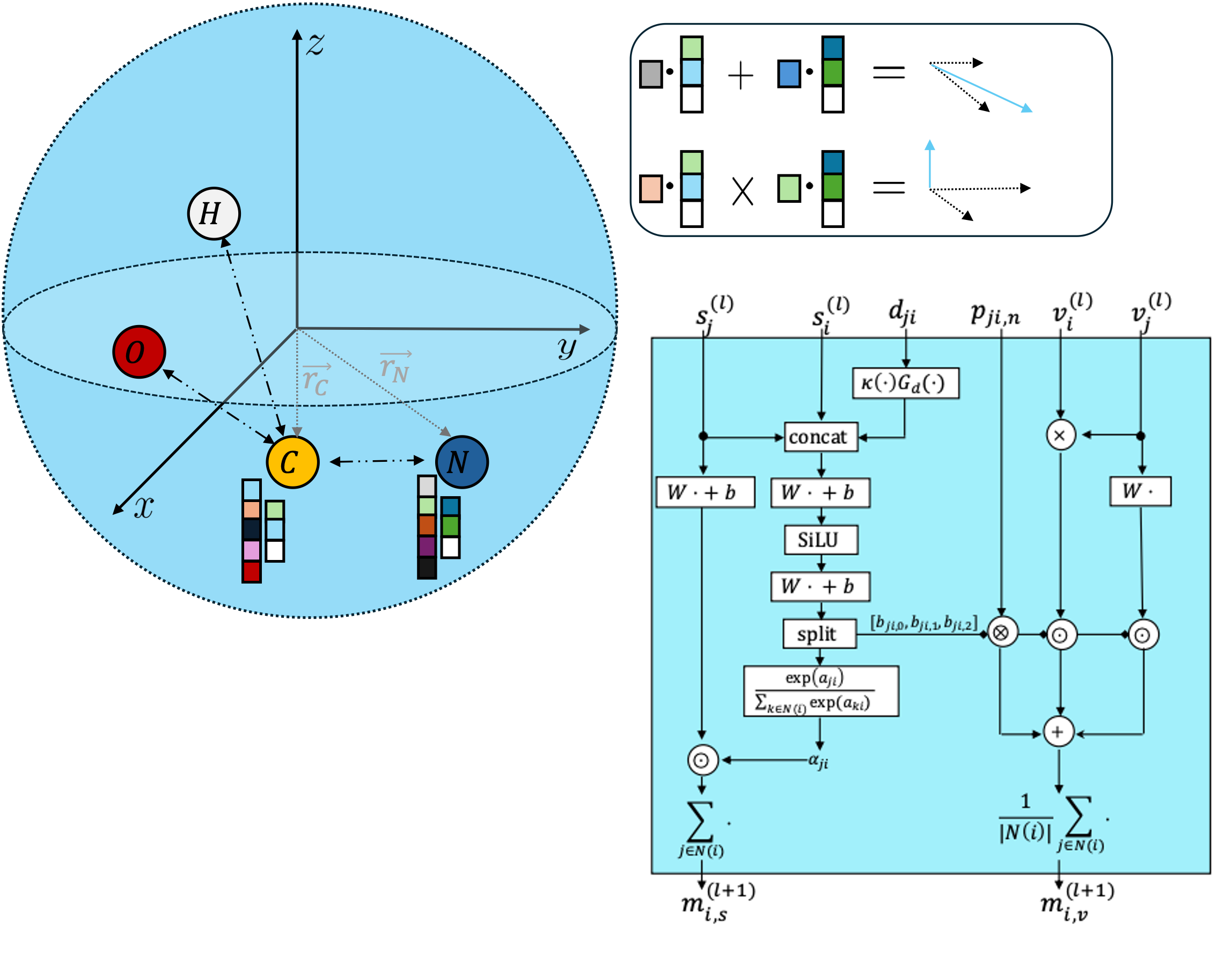 Representation Learning on Biomolecular Structures using
Representation Learning on Biomolecular Structures using
Equivariant Graph AttentionTuan Le*, Frank Noé, and Djork-Arné ClevertIn Learning on Graphs Conference May 2022Learning and reasoning about 3D molecular structures with varying size is an emerging and important challenge in machine learning and especially in the development of biotherapeutics. Equivariant Graph Neural Networks (GNNs) can simultaneously leverage the geometric and relational detail of the problem domain and are known to learn expressive representations through the propagation of information between nodes leveraging higher-order representations to faithfully express the geometry of the data, such as directionality in their intermediate layers. In this work, we propose an equivariant GNN that operates with Cartesian coordinates to incorporate directionality and we implement a novel attention mechanism, acting as a content and spatial dependent filter when propagating information between nodes. Our proposed message function processes vector features in a geometrically meaningful way by mixing existing vectors and creating new ones based on cross products. We demonstrate the efficacy of our architecture on accurately predicting properties of large biomolecules and show its computational advantage over recent methods which rely on irreducible representations by means of the spherical harmonics expansion.
-
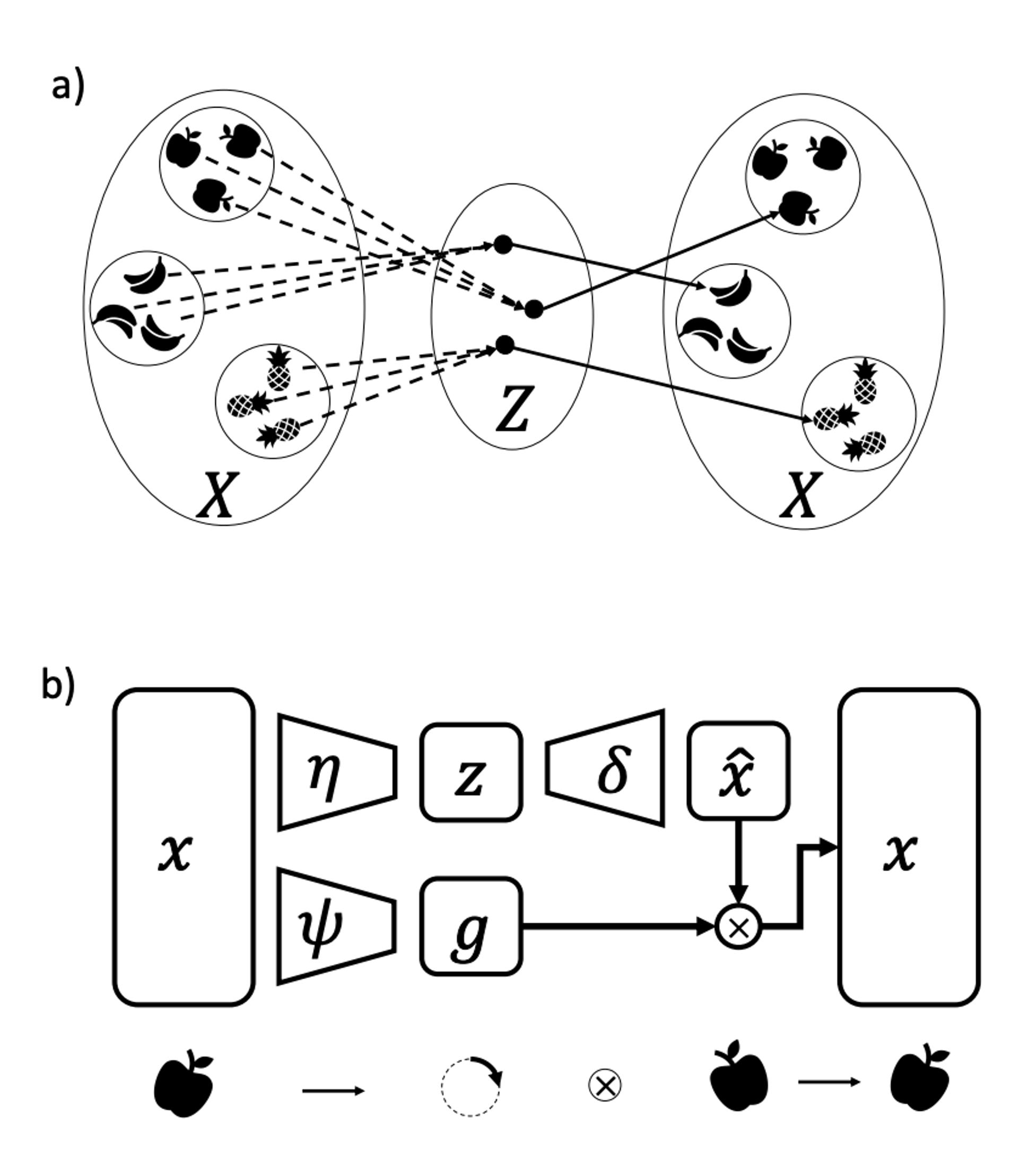 Unsupervised Learning of Group Invariant and Equivariant RepresentationsIn Advances in Neural Information Processing Systems May 2022
Unsupervised Learning of Group Invariant and Equivariant RepresentationsIn Advances in Neural Information Processing Systems May 2022Equivariant neural networks, whose hidden features transform according to representations of a group G acting on the data, exhibit training efficiency and an improved generalisation performance. In this work, we extend group invariant and equivariant representation learning to the field of unsupervised deep learning. We propose a general learning strategy based on an encoder-decoder framework in which the latent representation is separated in an invariant term and an equivariant group action component. The key idea is that the network learns to encode and decode data to and from a group-invariant representation by additionally learning to predict the appropriate group action to align input and output pose to solve the reconstruction task. We derive the necessary conditions on the equivariant encoder, and we present a construction valid for any G, both discrete and continuous. We describe explicitly our construction for rotations, translations and permutations. We test the validity and the robustness of our approach in a variety of experiments with diverse data types employing different network architectures.
2021
-
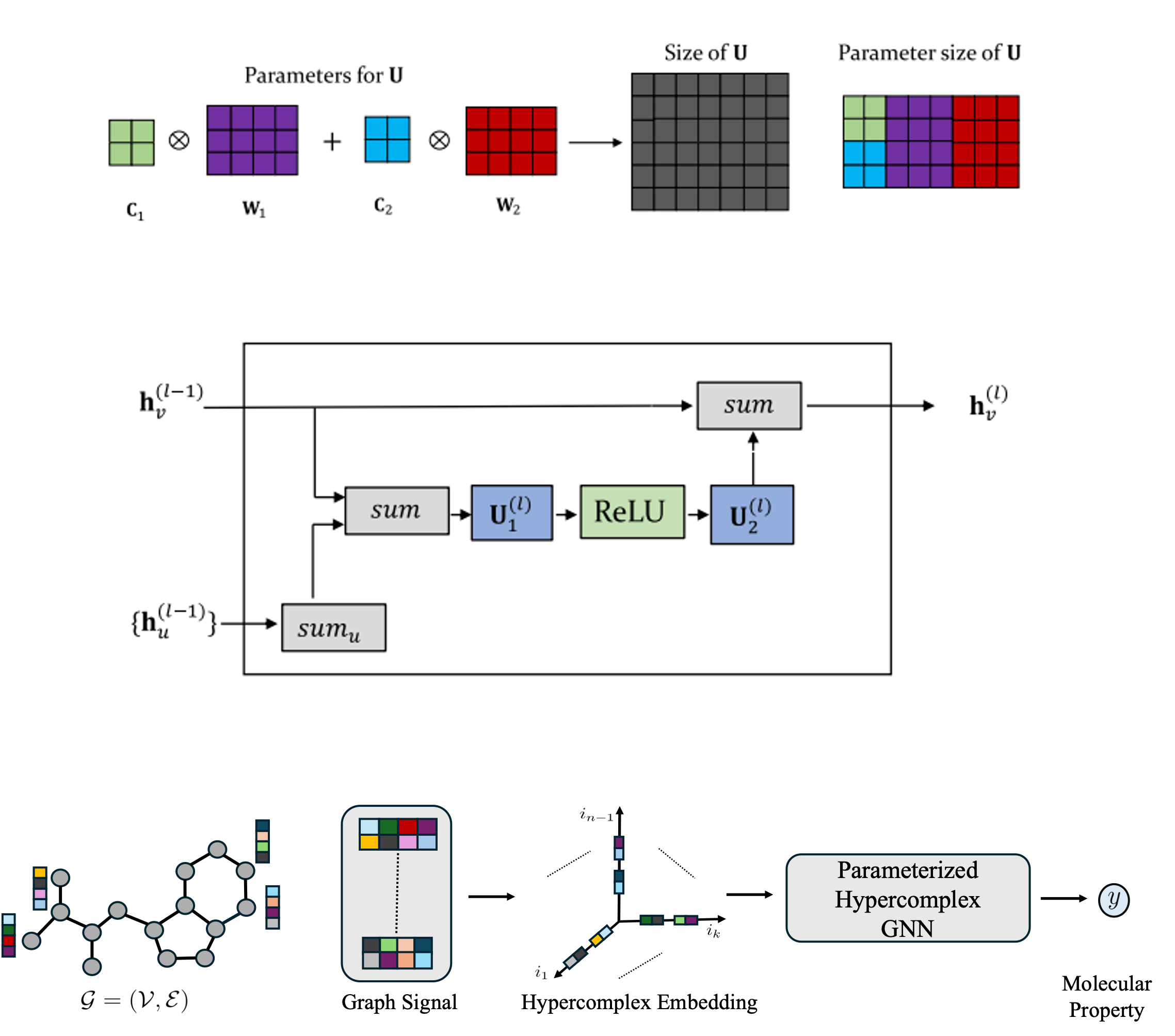 Parameterized Hypercomplex Graph Neural Networks for Graph ClassificationIn Artificial Neural Networks and Machine Learning – ICANN 2021 May 2021
Parameterized Hypercomplex Graph Neural Networks for Graph ClassificationIn Artificial Neural Networks and Machine Learning – ICANN 2021 May 2021Despite recent advances in representation learning in hypercomplex (HC) space, this subject is still vastly unexplored in the context of graphs. Motivated by the complex and quaternion algebras, which have been found in several contexts to enable effective representation learning that inherently incorporates a weight-sharing mechanism, we develop graph neural networks that leverage the properties of hypercomplex feature transformation. In particular, in our proposed class of models, the multiplication rule specifying the algebra itself is inferred from the data during training. Given a fixed model architecture, we present empirical evidence that our proposed model incorporates a regularization effect, alleviating the risk of overfitting. We also show that for fixed model capacity, our proposed method outperforms its corresponding real-formulated GNN, providing additional confirmation for the enhanced expressivity of HC embeddings. Finally, we test our proposed hypercomplex GNN on several open graph benchmark datasets and show that our models reach state-of-the-art performance while consuming a much lower memory footprint with 70% fewer parameters. Our implementations are available at https://github.com/bayer-science-for-a-better-life/phc-gnn.
-
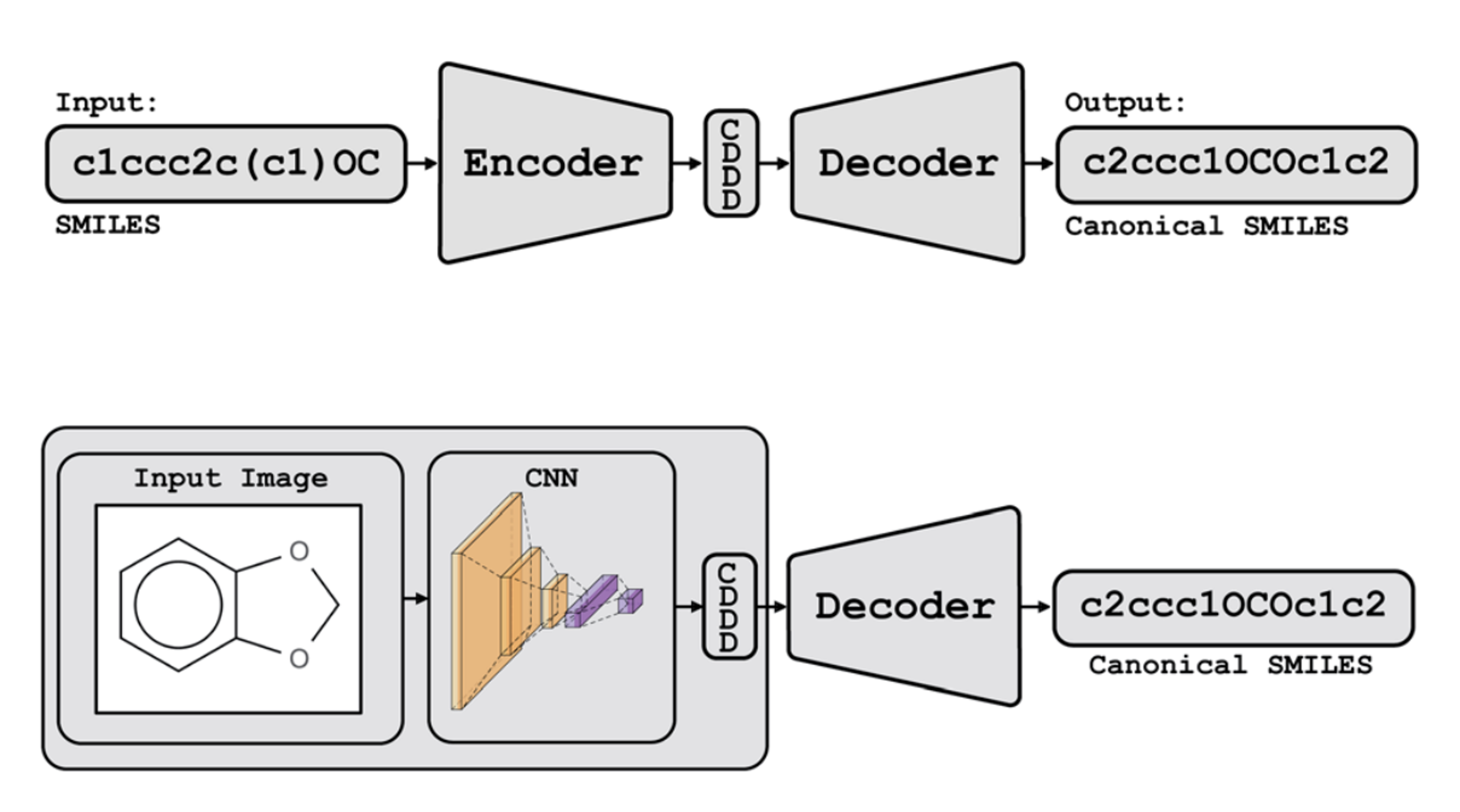 Img2Mol - Accurate SMILES Recognition from Molecular Graphical DepictionsChem. Sci. May 2021
Img2Mol - Accurate SMILES Recognition from Molecular Graphical DepictionsChem. Sci. May 2021"Automatic recognition of the molecular content of a molecule’s graphical depiction is an extremely challenging problem that remains largely unsolved despite decades of research. Recent advances in neural machine translation enable the auto-encoding of molecular structures in a continuous vector space of fixed size (latent representation) with low reconstruction errors. In this paper, we present a fast and accurate model combining a deep convolutional neural network learning from molecule depictions and a pre-trained decoder that translates the latent representation into the SMILES representation of the molecules. This combination allows to precisely infer a molecular structure from an image. Our rigorous evaluation show that Img2Mol is able to correctly translate up to 88% of the molecular depictions into their SMILES representation. A pretrained version of Img2Mol is made publicly available on GitHub for non-commercial users."
2020
-
 Going full hyper: hyperbolic and hypercomplex graph embeddings
Going full hyper: hyperbolic and hypercomplex graph embeddings
for ADMET modelingMay 2020We apply multitask learning in hyperbolic and hypercomplex spaces for predicting physico-chemical ADMET endpoints of small molecules. Our graph neural networks implementations show an increased overall predicting performance with respect to Euclidean-based methods. The performance gain of the quaternion model is especially accentuated in tasks with fewer data, strengthening the scope of multitask learning. In the hyperbolic approach, we experimentally observe that the network is making use of higher curvatures mainly in deeper layers, prompting us to explore hybrid networks, in which different layer geometries are combined.
-
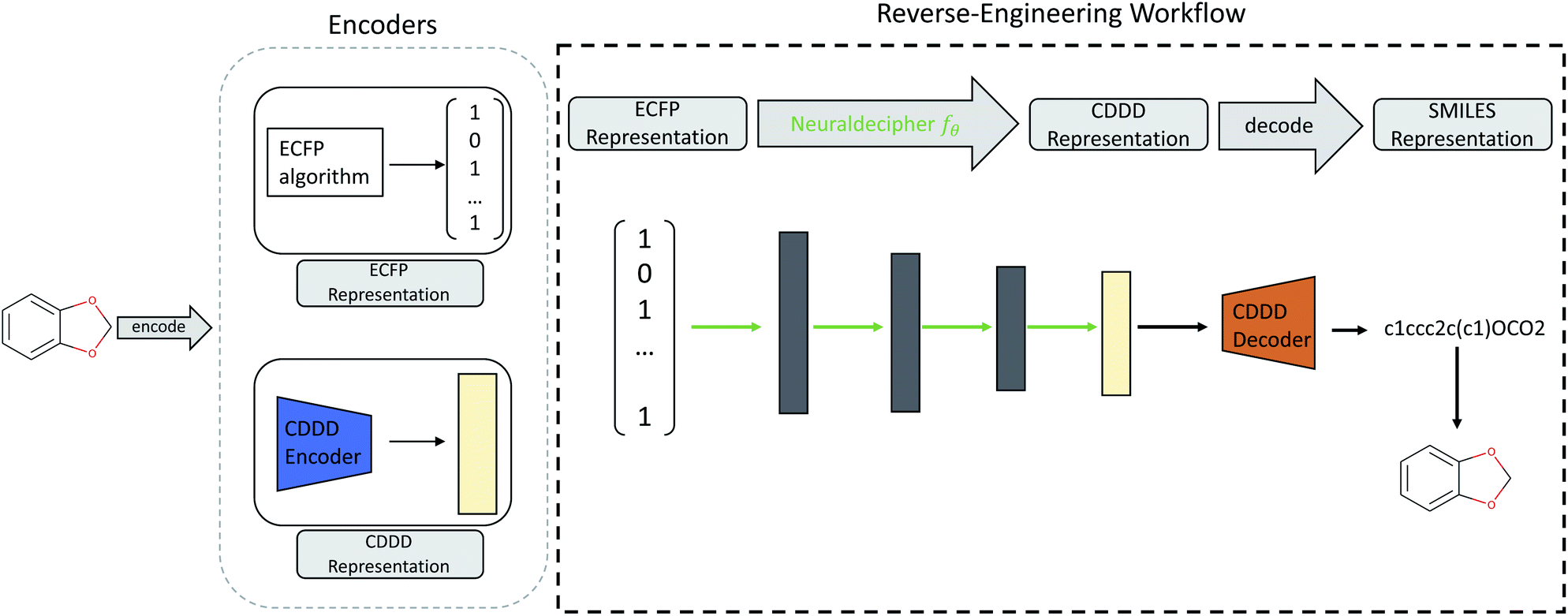 Neuraldecipher – reverse-engineering extended-connectivity fingerprints (ECFPs) to their molecular structuresChem. Sci. May 2020
Neuraldecipher – reverse-engineering extended-connectivity fingerprints (ECFPs) to their molecular structuresChem. Sci. May 2020Protecting molecular structures from disclosure against external parties is of great relevance for industrial and private associations, such as pharmaceutical companies. Within the framework of external collaborations, it is common to exchange datasets by encoding the molecular structures into descriptors. Molecular fingerprints such as the extended-connectivity fingerprints (ECFPs) are frequently used for such an exchange, because they typically perform well on quantitative structure–activity relationship tasks. ECFPs are often considered to be non-invertible due to the way they are computed. In this paper, we present a fast reverse-engineering method to deduce the molecular structure given revealed ECFPs. Our method includes the Neuraldecipher, a neural network model that predicts a compact vector representation of compounds, given ECFPs. We then utilize another pre-trained model to retrieve the molecular structure as SMILES representation. We demonstrate that our method is able to reconstruct molecular structures to some extent, and improves, when ECFPs with larger fingerprint sizes are revealed. For example, given ECFP count vectors of length 4096, we are able to correctly deduce up to 69% of molecular structures on a validation set (112 K unique samples) with our method.